LC-MS okienko: Cielená kvantifikácia proteínov pomocou WiSIM-DIA a Orbitrap™ Fusion™ Lumos™ tribridného hmotnostného spektrometra
Komplexnosť biologických vzoriek a rozdielna koncentračná úroveň prítomných štruktúr proteínov kladie vysoké nároky na všetky analytické metódy a techniky. Hybridná hmotnostná spektrometria s vysokým rozlíšením a možnosťou rýchleho skenovania hrá nielen v proteomike esenciálnu úlohu pre dosiahnutie veľmi presných a reprodukovateľných dát. „Klasický“ data-dependentný prístup (DDA) je tak vďaka vyspelému inštrumentálnemu a softvérovému vybaveniu nahrádzaný data-independetnými analýzami (DIA).
Thermo Scientific™ Orbitrap Fusion™ Lumos™ Tribrid™ hybridný hmotnostný spektrometer je zložený z predradeného kvadrupólu, hmotnostného analyzátora na báze orbitálnej pasce (rozlišovacia schopnosť 500 000 pri 1.5 Hz), kolíznej cely a lineárnej iónovej pasce (˃20 kolízií -indukovaných disociáciou za 1 sekundu) (Q-OT-qIT) (Obr. 1). Unikátna tribridná architektúra tak dovoľuje analyzovať selektované ióny v orbitálnej pasci (SIM) s vysokou citlivosťou a paralelne vykonávať cielené MSn experimenty v iónovej pasci.
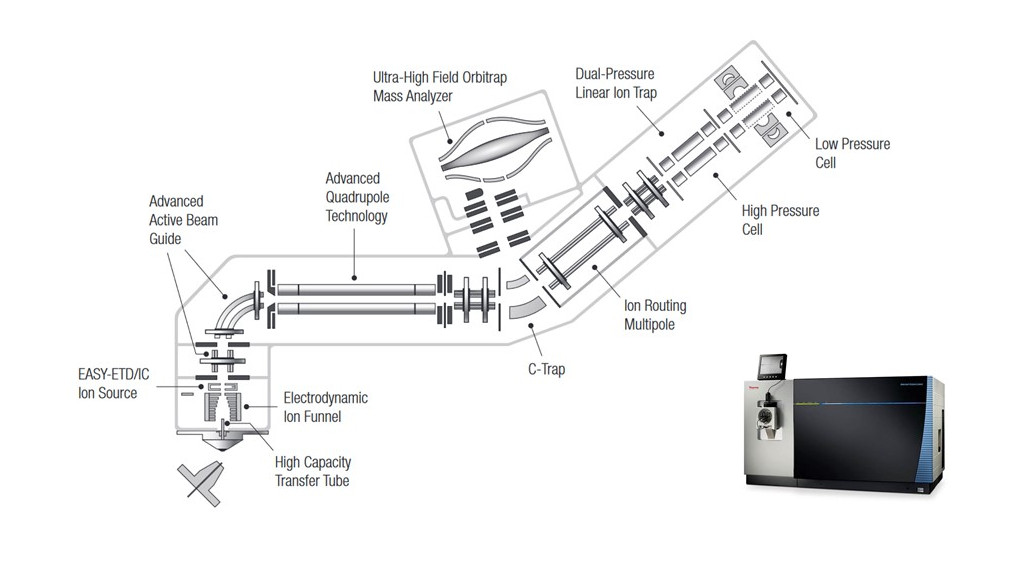
Obr. 1 - Thermo Scientific™ Orbitrap Fusion™ Lumos™ Tribrid™ hybridný hmotnostný spektrometer
Výhody tohto revolučného systému boli využité na tvorbu nového prístupu slúžiacemu k citlivej a zároveň robustnej kvantifikácii proteínov. Wide selected-ion-monitoring data independent analysis (WiSIM-DIA) je metóda zložená z troch po sebe nasledujúcich skenov dát s vysokou presnosťou a rozlíšením (240 000 FWHM, <3 ppm). Separátne skenovanie v celkovom rozsahu 400 – 1000 m/z zachováva rozsah full-scan experimentu, avšak výrazne zvyšuje citlivosť. Veľký rozsah m/z selektovaných iónov v prvom kroku zvyšuje pravdepodobnosť detekcie a následnej kvantifikácie nízko-abundantných proteínov. S každým SIM experimentom je súbežne v iónovej pasci izolovaných 17 MS/MS fragmentačných spektier prekurzorových iónov s „oknom“ 12 m/z , ktoré sú následne použité na identifikáciu peptidu na základe databázového porovnania. Toto usporiadanie tak plne pokrýva každý z troch skenov (Obr. 2).
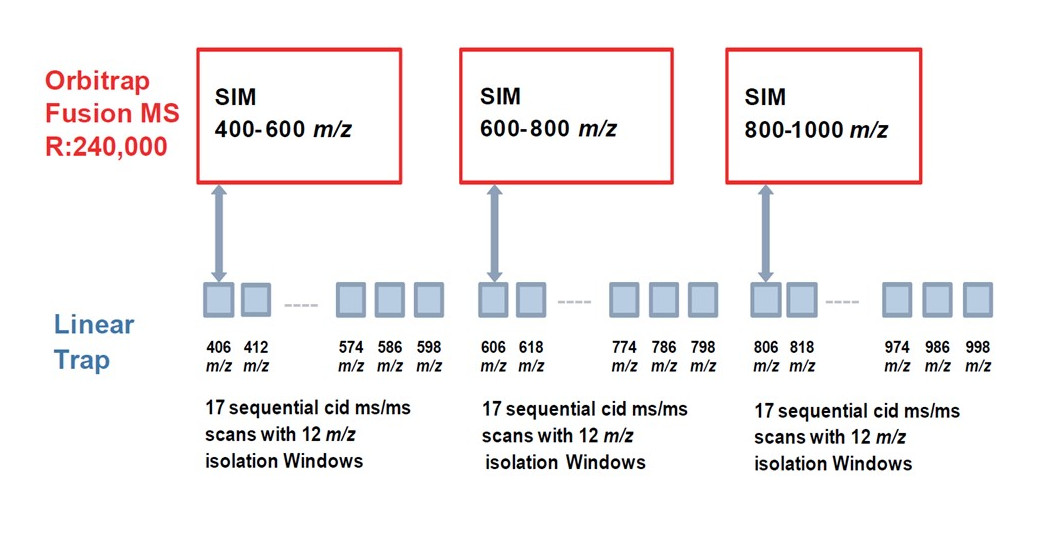
Obr. 2 - Schéma Wide selected-ion-monitoring data independent analysis (WiSIM-DIA) experimentu
Kvantifikácia proteínov je realizovaná na základe dát orbitálnej pasce s vysokým rozlíšením. MS/MS dáta z druhého kroku sú použité iba na konfirmačné účely. Koelúcia necielených proteínov tak neovplyvní kvantitatívne stanovenie, ako je tomu bežné v niektorých konvenčných DIA prístupoch, kde sú dáta získavané bez následnej konfirmácie.
Uvedený prístup vykazuje vďaka rozdeleniu do troch nadväzujúcich skenov s vysokým rozlíšením výrazne vyššie hodnoty pomeru signálu a šumu (S/N) oproti full-scan experimentu, napr. limit detekcie (LOD) bol na riedenej sérii 14 izotopovo značených peptidov v matrici E.coli stanovený na 10 amol/μl, päťbodová kalibrácia bola vykonaná v triplikátu, koeficient variácie (% CV) bol menší než 15%. V neposlednom rade - vierohodnosť identifikácie peptidov na základe databázového skríningu bola verifikovaná veľmi nízkou hodnotou p -value < 1 x 10-4 (Obr. 3).
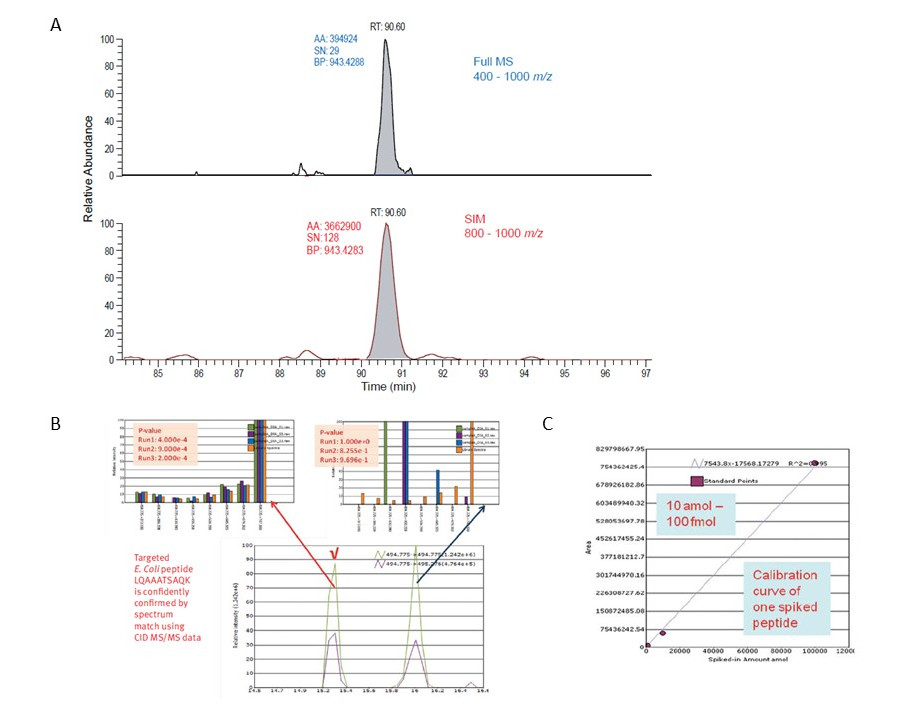
Obr. 3 – Porovnanie hodnoty signál/šum (S/N) v prípade full-scan a WiSIM-DIA usporiadania (A), výsledok databázového skríningu fragmentačných spektier 8 nejintenzívnejších iónov peptidu E.coli, pík v 16. min je kontaminant (B), kalibračná krivka jedného zo 14 izotopovo značených peptidov pridaných do matrice E.coli (C).
Pre ďalšie aplikácie a viac informácií: Application Note 600
Nenechajte si ujsť ďalšie zaujímavosti
- Thermo Scientific™ Dionex™ Inuvion™ IC - moderný systém iónovej chromatografie pre rutinné aj náročnejšie analýzy
- Thermo Scientific SMART chromatografické striekačky
- LC-MS/MS kvantifikácia voľných metanefrínov v ľudskej plazme pre klinický výskum
- Kvantifikácia ôsmich antimykotík v ľudskej plazme pomocou kvapalinovej chromatografie - tandemovej hmotnostnej spektrometrie pre klinický výskum
- Biopsia dychu pomocou technológie GC-Orbitrap – neinvazívny prístup k odhaleniu choroby?